近日,由我院人工智能科技专项参与资助的,中科院大连化学物理研究所催化基础国家重点实验室理论催化创新特区研究组(05T8组)肖建平研究员团队在电催化理论模拟方面取得新进展,提出了新的电化学恒电势模拟方法。相关研究以“Adaptive Electric Fields Embedded Electrochemical Barrier Calculations”为题,于近日发表在J. Phys. Chem. Lett.上。
电化学界面是变电子数的巨正则系综体系,在发生反应时,反应的能量性质依赖于电极电势。因为标准的第一性原理计算方法都是针对正则系综的,所以电化学计算的理论模拟比较困难。目前,电化学反应的模拟方法可以大体分为两类。一类是基于正则系综方法的后处理方法,比较典型的是计算氢电极模型和电容器模型,然而这些后处理方法无法描述反应过渡态随电势变化的现象。另一类方法是通过改变体系中电子数来控制体系的功函数(绝对电极电势)。虽然从概念上这类方法模拟的体系接近巨正则系综,但使用功函数作为电化学描述子时会存在能量的多值问题,例如反应能无法被功函数唯一确定,是功函数的多值函数等。
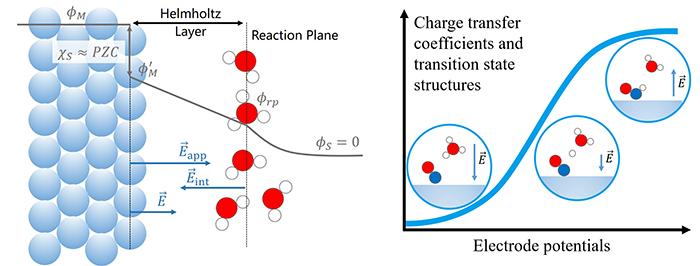
为了避免上述方法的问题,本工作中,团队提出了把Helmholtz层中电场强度E作为控制变量的恒电势模拟方法(electric field controlling constant potential, EFC-CP)。在该方法中,首先E和电极电势φM通过经典双电层模型(Stern模型)建立了一一对应关系。在给定电极电势φM时,在体系上施加外电场E。如果表面因为电解质的存在而带电,需要减去表面电荷导致的电场。在该方法中,氢还原反应反应自由能为0的电势被定义为内部可逆氢电极(IRHE)电势。当以IRHE作为电势参考时,EFC-CP计算得到的反应能跟计算氢电极模型接近。并且,在密度泛函理论计算的误差范围内,反应能计算结果与搭建的模型无关,避免了反应能多值问题。该团队进一步用EFC-CP方法研究了Ag电极表面一些电化学基元反应,讨论了电荷转移系数和过渡态结构随电极电势的变化。
该工作得到了我院人工智能科技专项一期项目“人工智能赋能多相催化探索研究”的资助,项目负责人是大连化物所肖建平研究员,项目编号为DNL-YL A202205。
原文链接:http://www.dicp.cas.cn/xwdt/kyjz/202301/t20230119_6601646.html
论文链接:https://doi.org/10.1021/acs.jpclett.2c03588



